Genome dynamics and instability
This team belongs to the UMR 9019 Genome Integrity and Cancers.
Research topics
Genome dynamics and stability
The Genome dynamics and stability team aims to understand how alterations in genome metabolism and stability impact tumorigenesis, cancer progression, treatment choice, and aging.
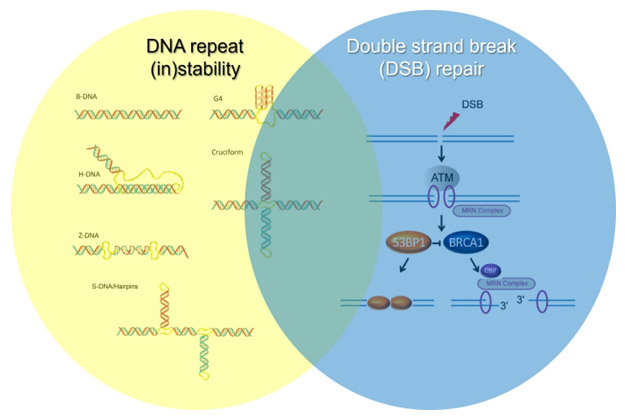
More specifically, the group studies the mechanisms controlling the (in)stability of repetitive genomic regions and the link between repeat instability, the formation of alternative DNA structures, and targeted cancer therapies. Additionally, the team seeks to understand how the choice between different double-strand break (DSB) repair pathways influences the balance between tumorigenesis and aging, and how DSB repair is connected to DNA repeat instability.
The team's projects employ a wide range of experimental approaches, including mouse and patient-derived cellular models, whole-genome sequencing using both short- and long-read technologies, functional genomic assays (e.g., CRISPR screens, ChIP-seq, END-seq, RNA-seq), and big data analytics.
Mechanisms of Formation and Targeting of DNA Triplexes in Cancer
Approximately 3% of the human genome consists of simple tandem DNA repeats, comprising over one million sequences capable of forming alternative (non-B) DNA structures, such as cruciforms, triplexes (H-DNA), and guanine quadruplexes (G4-DNA). These structures contribute to genome instability and are enriched in cancer, a feature now being exploited for therapeutic purposes.
Our research has shown that cancer cells exhibit high levels of DNA triplexes(H-DNA)—alternative DNA structures formed during replication that promote genome instability. Our group aims to:
- Understand the mechanisms driving DNA triplex formation in cancer cells;
- Identify the tumor types and biological contexts in which DNA triplexes can be therapeutically targeted; and
- Use or develop small molecules that bind and stabilize DNA triplexes to selectively eliminate cancer cells.
MSI Cancers: Repeat Instability and Novel Therapeutic Interventions
Microsatellite instability (MSI) is a mutational signature associated with repetitive DNA sequences and results from defective mismatch repair (dMMR). MSI prevalence varies by cancer type, reaching approximately 31% in endometrial carcinomas and over 10% in colon and stomach adenocarcinomas. In recent years, dMMR/MSI tumors have shown strong responses to immune checkpoint inhibitors, expanding treatment options and improving patient survival. However, resistance to these therapies remains a significant challenge, underscoring the need for alternative treatments.
We aim to identify additional vulnerabilities in MSI tumors to establish therapeutic strategies that could make dMMR/MSI tumors fully treatable.
Recent preclinical studies have demonstrated that MSI tumors are particularly sensitive to inactivation of the DNA helicase WRN, which is thought to resolve alternative DNA structures arising from repeat expansions — features best characterized via 3rd-generation (long-read) sequencing. WRN inhibitors are under development, and clinical trials targeting WRN in MSI tumors are underway.
In our laboratory, we aim to:
- Develop sequencing protocols to characterize repeat instability in dMMR/MSI tumors;
- Elucidate the mechanisms underlying repeat instability in MSI tumors; and
- Develop rationales and diagnostic tools to better identify patients who may benefit from WRN inhibitor therapies.
Double-Strand Break Repair Pathway Choice and Its Role in Tumorigenesis
Double-strand breaks (DSBs) are among the most toxic forms of DNA damage. Homologous recombination (HR) is a key pathway for DSB repair, particularly during the S-G2 phases of the cell cycle, where it ensures faithful replication fork progression. Homologous recombination deficiency (HRD) is found in multiple cancer types. Notably, germline inactivating mutations in BRCA1 or BRCA2 are associated with up to a 70% lifetime risk of breast cancer. For ovarian cancer, BRCA1 mutations confer a 44% risk, while BRCA2 mutations confer a 17% risk. HR gene inactivation can also cause developmental syndromes with childhood cancer predisposition, such as Fanconi anemia resulting from homozygous BRCA1 (FANC-R) or BRCA2 (FANCD1) mutations.
HRD is not limited to inherited mutations—it can also result from a spectrum of somatic mutations and epigenetic silencing events. Despite its prevalence and clinical significance, the mechanisms by which HRD initiates and promotes cancer are not fully understood.
Our research addresses the following questions:
- What mechanisms govern DSB repair pathway choice?
- Does HR deficiency intrinsically lead to tumorigenesis, or does it do so by promoting alternative, error-prone repair mechanisms?
- How are inflammation and aging linked to defective HR-mediated DNA repair?